
- extracting RNA secondary structure from a PDB or PDBx/mmCIF file,
- converting CT to BPSEQ and vice versa,
- converting CT and BPSEQ to dot-bracket,
- encoding extracted RNA secondary structure in dot-bracket, CT and BPSEQ,
- creating a visualization of RNA secondary structure,
- finding multiple 2D structures from a number of 3D structure analyses.
- 3D → (....), in which the secondary structure is extracted from a PDB or PDBx/mmCIF file,
- 2D → (....), where the secondary structure topology is derived from a list of base pairs,
- (....) → image, where the secondary structure is visualized basing on a provided topology,
- 3D → multi 2D, where multiple secondary structures are found.
3D → (....) scenario
- Step 1
-
In the first step of this scenario, a user should upload the PDB or PDBx/mmCIF file with RNA tertiary
structure. The file can be uploaded either directly from a local drive (use "Browse" button to browse
through the local repositories) or from Protein Data Bank. In the second case a user should enter PDB
identifier into the edit box, and click the "Get" button. The associated PDB or PDBx/mmCIF file is
automatically downloaded from Protein Data Bank and made ready for processing by the application.
There are 5 example PDB and PDBx/mmCIF files stored in the system and ready for processing. They enable new users an easy start with RNApdbee. Uploaded data can be viewed in the textarea after clicking "Show file contents" button, and edited before further processing.
- Step 2
-
In the second step, an application used to identify base pairs in
the uploaded structure should be selected from four available
programs: 3DNA/DSSR (default), RNAView, MC-Annotate, FR3D.
3DNA/DSSR can be selected with additional option "Analyse helices".
Citing 3DNA/DSSR documentation: "a helix is defined by
base-stacking interactions, regardless of bp type and backbone
connectivity". Therefore, when this option is set in RNApdbee, the
resulting 2D structure (both in text and visualization) will
contain pairs which in 3D structure form such defined helix, even
if the nucleotides from opposite strands are not constituting
canonical base pairs.
Additionally, if one wants to have non-canonical base pairs
annotated in the output (either in text and visualization, or
visualization only), an appropriate option should be checked. By
default, RNApdbee output representations contain only canonical
base pairs, while non-canonical ones are included in a separate
list only. In this step, one can also decide to remove isolated
base pairs from the result.
- Step 3
-
In the third step, an algorithm to resolve and encode secondary structure topology should be selected. There
are five available options to choose from: Hybrid Algorithm (default), Dynamic Programming, Elimination
Min-Gain, Elimination Max-Conflicts, First-Come-First-Served. Each algorithm is given a list of base pairs
(identified by a tool from previous step) and proceeds to encode the secondary structure topology, which is
a difficult task for highly complex and pseudoknotted RNA structures. Hybrid Algorithm performs an
exhaustive search for small subproblems and random walk for larger ones. The class of Elimination algorithms
works iteratively by selecting certain base pairs to be encoded as pseudoknots. The selection criterion is
different in the Min-Gain and Max-Conflicts variants yielding differences in results. In Min-Gain, base
pairs are analysed from the perspective of stem size they are part of and adjacent stem sizes. In
Max-Conflicts, the number of adjacent stems is taken into account. The Dynamic Programming works on the same
principle as Elimination algorithms, but the selection criterion is such as to optimize the global solution.
The First-Come-First-Served algorithm is a simple heuristic encoding the topology from 5' to 3' ends.
- Step 4
-
RNApdbee automatically finds secondary structure elements within the result. In this step, user can decide
to treat pseudoknots as either paired (default) or unpaired residues. With the default option selected,
residues encoded as pseudoknots will be allowed to form stems and terminating base pairs of loops. When the
other option is set, the same residues will be part of single strands and inside of loops.
- Step 5
-
Next, the user should decide whether the visualization is to be generated at the output, together with the
textual representation of the resulting secondary structure. In order to get an image, one should select one
of three available procedures: VARNA-based (default), PseudoViewer-based or R-chie-based procedure.
Otherwise, 'No image' option must be selected. The first two procedures generate a classical visualization
of secondary structure, while the last one produces an arc diagram. If the selected procedure fails to
generate an image, the alternative procedure is automatically run.
- Step 6
- To start secondary structure extraction, the "Run" button should be clicked. This causes an immediate display of the results page with the secondary structure encoded in extended dot-bracket notation, BPSEQ and CT, listing of non-canonical base pairs and other RNA tertiary contacts with their classification, identified structural elements, and visualization (if previously requested). The results can be saved to a local drive. Moreover, the same input data can be processed again with the other set of options.
2D → (....) scenario
- Step 1
-
In the first step of this scenario, the user should upload the BPSEQ or CT file from local repository (use
"Browse" button to select a file from the required folder). There are also 3 example files in BPSEQ format
and 3 example files in CT format available for upload. Uploaded data can be viewed in the textarea after
clicking "Show file contents" button, and edited before further processing.
- Step 2
-
In this step, the user can select how should isolated base pairs be treated. By default, they are included
in the result just as any other pairing. In an isolated base pair, in the close vicinity of both residues
there are no other interactions to stabilise the pairing. Therefore, user can decide to treat it as unsure
or unstable and remove it from the result.
- Step 3
-
In the third step, an algorithm to resolve and encode secondary structure topology should be selected. There
are five available options to choose from: Hybrid Algorithm (default), Dynamic Programming, Elimination
Min-Gain, Elimination Max-Conflicts, First-Come-First-Served. Each algorithm is given a list of base pairs
(identified by a tool from previous step) and proceeds to encode the secondary structure topology, which is
a difficult task for highly complex and pseudoknotted RNA structures. Hybrid Algorithm performs an
exhaustive search for small subproblems and random walk for larger ones. The class of Elimination algorithms
works iteratively by selecting certain base pairs to be encoded as pseudoknots. The selection criterion is
different in the Min-Gain and Max-Conflicts variants yielding differences in results. In Min-Gain, base
pairs are analysed from the perspective of stem size they are part of and adjacent stem sizes. In
Max-Conflicts, the number of adjacent stems is taken into account. The Dynamic Programming works on the same
principle as Elimination algorithms, but the selection criterion is such as to optimize the global solution.
The First-Come-First-Served algorithm is a simple heuristic encoding the topology from 5' to 3' ends.
- Step 4
-
RNApdbee automatically finds secondary structure elements within the result. In this step, user can decide
to treat pseudoknots as either paired (default) or unpaired residues. With the default option selected,
residues encoded as pseudoknots will be allowed to form stems and terminating base pairs of loops. When the
other option is set, the same residues will be part of single strands and inside of loops.
- Step 5
-
Next, the user should decide whether the visualization is to be generated at the output, together with the
textual representation of the resulting secondary structure. In order to get an image, one should select one
of three available procedures: VARNA-based (default), PseudoViewer-based or R-chie-based procedure.
Otherwise, 'No image' option must be selected. The first two procedures generate a classical visualization
of secondary structure, while the last one produces an arc diagram. If the selected procedure fails to
generate an image, the alternative procedure is automatically run.
- Step 6
- To start secondary structure processing, the "Run" button should be clicked. This causes an immediate display of the results page with the secondary structure encoded in extended dot-bracket notation, BPSEQ and CT, identified structural elements, and visualization (if requested). The results can be saved to a local drive. Moreover, the same input data can be processed again with the other set of options.
(....) → image
- Step 1
-
In the first step of this scenario, the user should upload the dot-bracket file from local repository (use
"Browse" button to select a file from the required folder). There are also 3 example files in dot-bracket
format available for upload. Uploaded data can be viewed in the textarea after clicking "Show file contents"
button, and edited before further processing.
- Step 2
-
RNApdbee automatically finds secondary structure elements within the result. In this step, user can decide
to treat pseudoknots as either paired (default) or unpaired residues. With the default option selected,
residues encoded as pseudoknots will be allowed to form stems and terminating base pairs of loops. When the
other option is set, the same residues will be part of single strands and inside of loops.
- Step 3
-
Next, the user should select one of three available procedures: VARNA-based (default), PseudoViewer-based or
R-chie-based procedure. The first two procedures generate a classical visualization of secondary structure,
while the last one produces an arc diagram. If the selected procedure fails to generate an image, the
alternative procedure is automatically run.
- Step 4
- To start visualization of the secondary structure topology, the "Run" button should be clicked. This causes an immediate display of the results page with the secondary structure encoded in extended dot-bracket notation, BPSEQ and CT, identifies structural elements, and visualization. The results can be saved to a local drive. Moreover, the same input data can be processed again with the other set of options.
3D → multi 2D
- Step 1
-
In the first step of this scenario, a user should upload the PDB or PDBx/mmCIF file with RNA tertiary
structure. The file can be uploaded either directly from a local drive (use "Browse" button to browse
through the local repositories) or from Protein Data Bank. In the second case a user should enter PDB
identifier into the edit box, and click the "Get" button. The associated PDB or PDBx/mmCIF file is
automatically downloaded from Protein Data Bank and made ready for processing by the application.
There are 5 example PDB and PDBx/mmCIF files stored in the system and ready for processing. They enable new users an easy start with RNApdbee. Uploaded data can be viewed in the textarea after clicking "Show file contents" button, and edited before further processing.
- Step 2
-
In the second step, user can configure parameters of analysis. The first option (off by default) if selected
would allow non-canonical base pairs to be included in the secondary structure topology. The second option
(off by default) if selected would remove any isolated base pairs from the result.
- Step 3
- Next, the user should decide whether the visualization is to be generated at the output, together with the
textual representation of the resulting secondary structure. In order to get an image, one should select
one of three available procedures: VARNA-based (default), PseudoViewer-based or R-chie-based procedure.
Otherwise, 'No image' option must be selected. The first two procedures generate a classical visualization
of secondary structure, while the last one produces an arc diagram. If the selected procedure fails to
generate an image, the alternative procedure is automatically run.
- Step 4
- To start the analysis, the "Run" button should be clicked. This causes an immediate display of the results page with possibly several secondary structures encoded in BPSEQ and CT format. Each entry in the result page is encoded by up to several different dot-bracket structures, each accompanied with an image (if requested). The results can be saved to a local drive.
Base-pair identification
RNAView
RNAView identifies and classifies the types of base pairs and basic RNA motifs such as loops and bulges that are formed in nucleic-acid structures. It provides the implementation of edge-to-edge hydrogen bonding interactions according to Leontis/Westhof nomenclature . The program allows also for identification of tertiary interactions and visualization of 2D diagrams of RNA secondary topology in Postscript, VRML or RNAML format. RNAView can be downloaded from http://ndbserver.rutgers.edu/ndbmodule/services/download/rnaview.html and used after successful installation.
MC-Annotate
MC-Annotate provides a structural graph which encodes geometric information based on atomic coordinates and torsion angles. In general, the structural graph represents the description of every nucleotide conformation (based on sugar puckering modes and nitrogen base orientations around the glycosyl bond), base-base interactions (based on stacking and hydrogen bonding information) and pseudoknots identified in the input structure. MC-Annotate allows also for RNA motif recognition. It is available as a webserver at http://www-lbit.iro.umontreal.ca/mcannotate-simple/ .
3DNA/DSSR
3DNA/DSSR (DSSR: Dissecting the Spatial Structure of RNA) tool allows to characterize the geometric features of RNAs. It is provided as a command-line driven package, which was designed to analyse, reconstruct and visualise the three-dimensional nucleic acid structures. 3DNA/DSSR identifies base pairs of the input RNA structure taking into account modified and unmodified residues that form canonical (Watson-Crick) base pairs, non-canonical base pairs with at least one H-bond and non-pairing interactions (e.g. base stacking). Moreover, 3DNA/DSSR characterizes base pairs using both Leontis/Westhof and Saenger classifications. It detects triplets, higher-order base associations and pseudoknots. Additionally, 3DNA/DSSR provides RNA secondary structure in the dot-bracket notation. It is available as a web server at http://web.x3dna.org/dssr.
FR3D
FR3D is a suite of MATLAB programs to look for recurrent 3D motifs in RNA structure. This process includes classification of base pairs and stacking interactions. FR3D internally performs this operation using base-centered approach to analyse geometries of bases with respect to a common frame. The found base pairs are classified according to Leontis/Westhof notation. The web-accessible version of the tool is available as WebFR3D.
PseudoViewer
PseudoViewer allows for effective visualisation of large RNAs, also these including pseudoknots, as planar drawings. As an input, it accepts the sequence and the secondary structure data in the dot-bracket or paired format. At the output, a URL of the generated image file is returned. The visualization can be prepared in EPS, SVG, PNG, or GIF. PseudoViewer is available at http://pseudoviewer.inha.ac.kr/ .
VARNA (Visualization Applet for RNA)
VARNA is mainly an interactive software for drawing and editing RNA secondary structures. It supports several input file formats, including BPSEQ, CT and others. The main advantage of VARNA is its support for non-canonical base pairs (Leontis/Westhof nomenclature) and pseudoknots (first extracted maximal planar subset of canonical base pairs is a scaffold for the rest of the base pairings). The output visualisation can be extracted in vector and bitmap picture formats including EPS, SVG, XFIG, JPG, or PNG. VARNA is available as the lightweight applet and swing component at http://varna.lri.fr/.
R-chie
R-chie is a tool for generating different types of arc diagrams. It supports visualization of multiple sequence alignments and incorporation of co-variance information into the image. R-chie can read inputs in various formats including BPSEQ, CT and dot-bracket, also these containing higher order pseudoknots. It is highly configurable and allows to save output as PNG or PDF files. The webserver is available at http://www.e-rna.org/r-chie.
Pseudoknot annotation scripts
Graphical images of RNA secondary structure generated by PseudoViewer and VARNA are post-processed by the RNApdbee scripts. Their aim is to annotate the orders of pseudoknot interactions in a graphical way. Different colours have been assigned to particular pseudoknot orders:
| Pseudoknot order | 1st | 2nd | 3rd | 4th | 5th | 6th | 7th | 8th |
| Text annotation (brackets & letter) | [] | {} | <> | Aa | Bb | Cc | Dd | Ee |
| Graphical annotation (colours) |
Scripts to annotate non-canonical interactions
Additional scripts post-process the VARNA and PseudoViewer images to display non-canonical interactions. VARNA visualization supports Leontis/Westhof classification and it is used during RNApdbee post-processing (see table below). In case of PseudoViewer, the non-canonical interactions are shown either as gray-filled circles (for regular bps) or coloured dashed lines (for pseudoknots, see table of colours above). Gray dashed lines are used to connect multiplet-involved residues and other pairs unrepresentable in text format.
| RNA base-base classification | Visualization symbol |
| cis Watson-Crick Watson-Crick |  |
| trans Watson-Crick Watson-Crick |  |
| cis Watson-Crick Hoogsteen |  |
| trans Watson-Crick Hoogsteen |  |
| cis Watson-Crick Sugar |  |
| trans Watson-Crick Sugar |  |
| cis Hoogsteen Watson-Crick |  |
| trans Hoogsteen Watson-Crick |  |
| cis Hoogsteen Hoogsteen |  |
| trans Hoogsteen Hoogsteen |  |
| cis Hoogsteen Sugar |  |
| trans Hoogsteen Sugar |  |
| cis Sugar Watson-Crick |  |
| trans Sugar Watson-Crick |  |
| cis Sugar Hoogsteen |  |
| trans Sugar Hoogsteen |  |
| cis Sugar Sugar |  |
| trans Sugar Sugar |  |
- PDB file stores various data concerning the three-dimensional structure of a
molecule, the experiment for structure determination, authors, etc. The
detailed description of this format is given
here. In RNApdbee, the
information about atom coordinates and missing or modified residues is
considered.
- PDBx/mmCIF is a dictionary-based storage format. The file contains well-formatted
entries containing 3D atom coordinates of a molecule as well as a set
of metadata related to the experiment of structure determination,
authors, etc. The detailed description of this format is given
here .
- BPSEQ file contains information about base pairs, stored in three columns: 1st
column contains the sequence position (starting at one), 2nd column contains
the base encoded in one-letter notation, 3rd column contains the pairing
base (if base from 2nd column is paired) or zero (if base from 2nd column is
unpaired).
- CT (connect) file is column based and contains the information about base pairs:
1st column specifies the sequence index (starting at one), 2nd column contains
the base in one-letter notation, 3rd and 4th columns specify additional indices
(the index of predecessor and successor of base in the chain; if one of them is
zero, it represents the terminal base in the chain), 5th column gives the
pairing base (if base from 2nd column is paired) or zero (if base from 2nd
column is unpaired) and 6th column corresponds to base number. Additionally, if
7th column appears and starts with a '#', the rest of the line contains a
comment.
- Dot-bracket notation is used to encode RNA secondary structure topology. Standard
dot-bracket encodes nested RNAs only. It is a string composed of dots
and brackets, where an unpaired nucleotide is represented as a dot
".", and a base pair is represented as a pair of opening (begin) and
closing (end) brackets, i.e. "(" and ")".
The extended dot-bracket is applied to represent knotted secondary structures: squared "[" and "]" brackets are used for lower-order structures, the curly brackets "{" and "}", angle brackets "<" and ">" and consecutive alphabet letters "A" and "a", "B" and "b", etc. represent higher orders and most complicated pseudoknots. Additionally, the minus sign "–" is used to encode an unidentified residue.
Example encoding of RNA secondary structure:
| BPSEQ format | CT format | dot-bracket notation | visualization |
| 1 G 8
2 G 7 3 C 0 4 A 0 5 U 0 6 U 0 7 C 2 8 C 1 |
8
1 G 0 2 8 1 2 G 1 3 7 2 3 C 2 4 0 3 4 A 3 5 0 4 5 U 4 6 0 5 6 U 5 7 0 6 7 C 6 8 2 7 |
GGCAUUCC
((....)) |
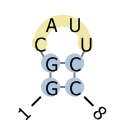 |
| Operating system | Recommended browser |
| Windows | Microsoft Internet Explorer (10 and later), Mozilla Firefox (4 and later), Opera (15 and later) or Google Chrome (19 and later) |
| Linux | Mozilla Firefox (4 and later), Opera (15 and later) or Google Chrome (19 and later) |
| macOS | Mozilla Firefox (4 and later), Opera (15 and later) or Google Chrome (19 and later) |
| Name | Version | Citation |
| 3DNA/DSSR | 1.7.6-2018mar22 | X.-J. Lu and W.K. Olson. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res, 31:5108–5121, 2003. |
| RNAView | N/A | H. Yang, F. Jossinet, N.B. Leontis, L. Chen, J. Westbrook, H. Berman, and E. Westhof. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res, 31:3450–3460, 2003. |
| MC-Annotate | 1.5 | H. Yang, F. Jossinet, N.B. Leontis, L. Chen, J. Westbrook, H. Berman, and E. Westhof. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res, 31:3450–3460, 2003. |
| FR3D | N/A | M. Sarver, C.L. Zirbel, J. Stombaugh, A. Mokdad, and N.B. Leontis. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J Math Biol, 56(1-2):215–252, 2007. |
| VARNA | 3.93-p1 | K. Darty, A. Denise, and Y. Ponty. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics, 25(15):1974–1975, 2009. |
| PseudoViewer | 3.0 | Y. Byun and K. Han. PseudoViewer: web application and web service for visualizing RNA pseudoknots and secondary structures. Nucleic Acids Res, 34(W1):W416–W422, 2006. |
| R-CHIE | 0.1.3 | D. Lai, J.R. Proctor, J.Y.A. Zhu, and I.M. Meyer. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res, 40(12):e95–e95, 2012. |
RNApdbee is using various maven-provided dependencies. The most important ones are presented below:
| Name | Version |
| org.biojava.biojava-structure | 5.0.0-alpha15 |
| org.springframework | 5.0.4.RELEASE |
| tiles-extras.tiles-extras | 3.0.8 |
| org.projectlombok.lombok | 1.16.20 |
| com.google.api-client.google-api-client | 1.23.0 |
- T. Zok, M. Antczak, M. Zurkowski, M. Popenda, J. Blazewicz, R.W. Adamiak, M. Szachniuk. RNApdbee 2.0: multifunctional tool for RNA structure annotation. Nucleic Acids Research 46(W1), 2018, W30-W35, (doi:10.1093/nar/gky314)
- M. Antczak, T. Zok, M. Popenda, P. Lukasiak, R.W. Adamiak, J. Blazewicz, M. Szachniuk. RNApdbee – a webserver to derive secondary structures from pdb files of knotted and unknotted RNAs. Nucleic Acids Research 42(W1), 2014, W368-W372, (doi:10.1093/nar/gku330)